In short:
XCrySDen is a
crystalline and molecular structure render program, with additional
capabilities of rendering contours, isosurfaces, Wigner-Seitz cells
(also Brillouin zone), Fermi surfaces and so on. Some of its
features are the following:
- widget with periodic table of elements:
- displaying molecular and crystalline structure in several
different display modes:
- displaying crystal and Wigner-Seitz cells:
- displaying smaller or bigger portion of crystal (multiplying
the unit cells)
- visualizing the forces as vectors:
- animations
- displaying contours and colorplanes
- displaying isosurfaces:

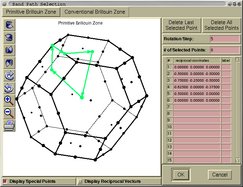
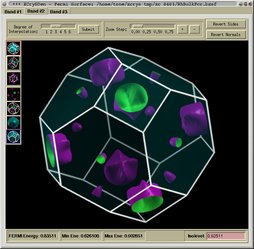
- reciprocal-space analysis:
+ selecting a k-path inside the Brillouin-zone
+ visualizing Fermi surfaces
- and more ...
This is the
XCrySDen
main window:
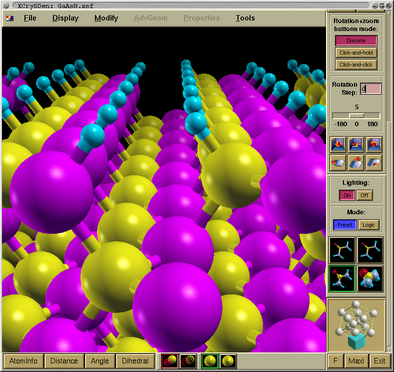
So what do we see:
- Top:
- A menu widget is at the top of the main window. Please
notice the Palette menu (small palette-like icon) at the
left of File menu. A background of the render window can
be set here to an arbitrary color.
- Middle:
- A big display area, where a portion of the GaAs surface with
adsorbed hydrogen is displayed, is a render window. Its
background color can be set via Palette menu (small
palette-like icon) at the left of File menu.
- Left:
- A toolbox panel is on the left side of the main window.
Please note a slider at its left. With it we can drag the panel up
and down, in order to reach all tools-widgets on the panel.
- Bottom:
- A measure buttons (AtomInfo, Distance, Angle,
Dihedral) are at the bottom of the main window. Here you can found how to measure distances,
angles and dihedrals. At a very right is the Exit
button.
Here you can found detailed description of
the
XCrySDen main
window. It was written for an old
XCrySDen's version 0.3. The main
window of the current version differs a bit, but nevertheless the
document is still useful.
- Rotation: press-and-hold left mouse
button and move the pointer
- Translation: press-and-hold middle mouse
button (or both buttons for two-button mouse) and move the
pointer
- Zoom (+): press-and-hold Shift + Left mouse
button and move the pointer up
- Zoom (-): press-and-hold Shift + Left mouse
button and move the pointer down
- Fullscreen mode: double-click the left mouse
button. To exit from the fullscreen mode perform another double
click with the left mouse button.
- Pop-up menu: press-and-hold the right mouse
button
XCrySDen has two levels
of display modes, these are so-called:
- Lighting-On mode yields fancy graphics with
shades. Example:
- Lighting-Off mode is very simple and fast, but
without shades. Example:
WARNING: Lighting-Off mode can display only
structure, crystal cages and nothing else !!!
NOTE: I used to receive several dozens Emails per year
claiming: "not able to render isosurface". The reason is
always the Lighting-Off mode.
When rendering an isosurface or some other complex object (i.e.
isosurface) switch to
Lighting-On mode, otherwise it will
not appear !!!
If your graphics card isn't fast, then the rotation will be quite
slow when rendering in
Lighting-On mode a very complex
object (hundreds of atoms, isosurfaces, etc.). In such a case,
simply switch to
Lighting-Off mode (this will turn off the
complexity) and rotate, translate, zoom and so on until you are
satisfied with the setting. Then switch back to
Lighting-On mode (this will turn on the isosurface) and
you will have the desired picture !!!
After the installation of the
XCrySDen program (the
Installation instructions can be found
here) we are ready to run
XCrySDen for the first time. The
name of
XCrySDen
executable is
xcrysden. I suggest that you start
exploring the program capabilities by running the provided examples
as explained below.
XCrySDen distribution
provides several example files. They can be loaded via
File-->XCrySDen Examples ... cascade menu.
There one can choose among different provided files in several
supported formats. After loading the structure, start to explore
the functionality of the toolbox panel buttons. The panel is
located on the left side of the main window. Try also to measure
some distances or angles by clicking the corresponding measure
buttons located at the bottom of the main window (the
HOWTO
Measure instructions can be found
here).
There are two ways to load the structure into
XCrySDen: (i) trough
File-->Open * menus (where "*" is a
wildcard standing for any text), or (ii) via command line options.
Execute:
xcrysden --help to display all supported
options.
XCrySDen
supports well-known
XYZ and
PDB file formats. In
the latter case only a subset of the file is read, namely the ATOMS
section, while all others are ignored. But these file formats do
not have support for crystal structures. Therefore
XCrySDen has its own file format,
which is called
XCrySDen
Structure
File (XSF)
format. Many other formats (like WIEN2k struct file or PWscf input
file) are supported through conversion to the XSF format.
Here you can find the
XSF format
specification.
The following file formats are supported in
XCrySDen:
- Molecular structure formats:
-
- XYZ
- PDB
- GAUSSIAN Z-Matrix file (requires BABEL program)
- GAUSSIAN output file
- Crystal structure formats:
-
- XSF
(XCrySDen Structure File)
- CRYSTAL files:
+ input file
+ unit 9 (i.e. properties file)
- WIEN2k struct file
- PWscf files:
+ input file
+ output file (structural optimization run
only)
- FHI98MD files:
+ inp.ini file
+ coord.out file
In addition, Jens Kunstmann has written an XSF-converting tool
xsfConvert.
Currently it can convert the data from the
TB-LMTO-ASA
and
VASP programs
to the XSF format.
The measurement of angles, distances and dihedrals can be easily
achieved by pressing corresponding
measure button. These buttons are located at the
bottom of
XCrySDen main
window. After pressing one of these buttons, the
measure
toplevel window will pop up. Please note that while in
measure
mode most of the functionality of
XCrySDen is frozen (for example: the
display-mode
cannot be changed). But we
can rotate
the displayed structure, and this feature is quite useful for
complex structures. If we want to measure the distance between two
atoms, then after pressing the
Distance button, simply
click on two atoms. If some atom is selected then it is changed to
light green color and its coordinates appear in the
measure window. Below we can see a snapshot of the
measurement of the angle:
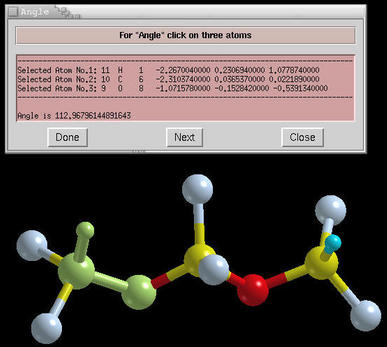
There are three buttons at the bottom of the
measure
window. The functionality of these buttons is:
- Done:
- calculates/determines the measured property
- Next:
- de-selects all selected atoms, and begins new selection
- Close:
- destroys the measure window and exits from selection
mode
XCrySDen menu widget is
located at the top of main window. There are the following
menu-buttons:
Palette, File, Display, Modify, AdvGeom,
Properties, and
Tools. They are shortly explained
below, but the detailed description can be found
here. I suggest you start by exploring various
entries in the
Display and
Modify menus.
 (Palette):
(Palette):- a background of the render window can be set here
- File:
- this menu is intended for Open, Save, Print, Exit
operations
- Display:
- this menu is intended for various "display" possibilities. It
consists of check- and radio-buttons.
- By clicking appropriate checkbuttons inside Display
menu one can render: Coordinates system (XYZ base vectors
are displayed at the bottom-left of the render window), Atomic
symbols, Crystal cells, Forces, Wigner-Seitz cells, Molecular surface.
- By clicking the appropriate radiobutton one can choose among:
- primitive or conventional setting of the unit cell
(Primitive cell Mode/Conventional Cell Mode
radiobuttons)
- criteria of rendering the ball and spacefill sizes, either
based on covalent or van der Waals radii
(radiobuttons in "Balls As ..." and "SpaceFill As
..." cascade menus)
- display style of crystal cells, either as lines or rods
(radiobuttons in "Crystal Cells As ..." cascade menu)
- crystal unit type ("Unit of Repetition ..." cascade
menu). One can choose among Unit Cell and
Translational asymmetric unit. What do we mean by this?
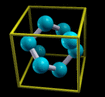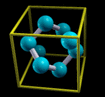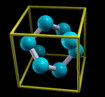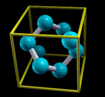
Simply rendering just atoms belonging to the zero reference unit
cell would not appear very nice. Why? Let us take for example an
fcc unit cell. The atoms belonging to this cell have the
following crystal (i.e. fractional) coordinates: (0,0,0);
(1/2,1/2,0); (1/2,0,1/2); (0,1/2,1/2). If solely this atoms are
rendered they appear as shown on the left of the below figure.
Instead, what one really wants is the shown on the right of the
figure:


The content displayed on the left of the figure is called
Translational asymmetric unit, while the one on the right
is called "nicely cut unit cell" or shortly Unit
Cell.
- Modify:
- this menu is dedicated to: (i) modifying miscellaneous items,
like various colors (for example: atom and cell
colors), atomic radii, widths of bonds and cells, ball-factor,
tessellation-factor, ...; (ii) setting the display of
forces, Wigner-Seitz cells, molecular surfaces,
animation. Read more ...
- AdvGeom:
- this menu is mented solely for CRYSTAL GUI. Here one can
manipulate an existing structure by various procedures. Among
supported options are: (i) cutting a slab out of a crystal, (ii)
generating a supercell, (iii) adding, removing and substituting
atoms, ...
- Properties:
- this menu is mented solely for CRYSTAL GUI. Here one can
perform some property analysis. Among supported options are the
plotting of (i) band widths, (ii) density-of-states, (iii) band
structure (one can select th k-path inside Brillouin zone
graphically), (iv) electronic densities and electrostatic
potentials as 2D contours or 3D isosurfaces.
- Tools: (in old versions this menu was called
Analysis)
- A few useful tools can be found here. This includes: Color
Scheme, Data-Grid, k-path Selection and Periodic Table of
Elements.

![[Figure]](img/xcrysden-picture-small-new.jpg)