NOTE:with the new
development of
XCrySDen
this document has became a bit obsolete. Several new menu items
were added and a few were rearanged since this document was
written, nevertheless it should still be useful.
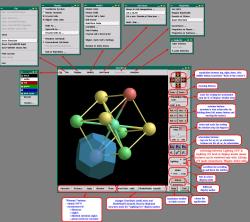
Below is a link to an interactive snapshot of
XCrySDen main window with its menus
as they appeared in an old version 0.3. The appearence of the main
window and menus in the current version differs a bit, nevertheless
the snapshot is still useful. However, the description of the menus
that can be found here below refers to the new version.

Changes the background color of render window. Custom background
color can be specified as well (via
Custom ...
entry).
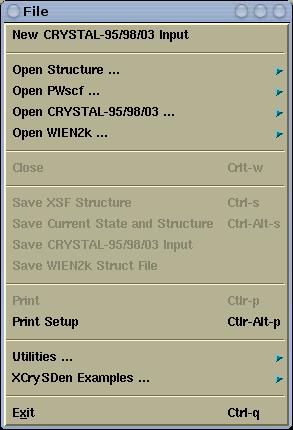
File menu has the followins items:
Creates new CRYSTAL-95/98/03 input file (just standard geometry
input and
geometry manipulation keyword sections (refer to
CRYSTAL-95/98/03 User manual). User will be able to construct new
structure from scratch.
Molecular or crystalline structure will be read from file and
displayed. Here one can open the following formats:
- Crystalline structures files:
- XCrySDen files:
- XSF (XCrySDen Structure File)
- AXSF (Animation XCrySDen Structure
File)
- BXSF (i.e. Fermi Surface
Files)
- XCrySDen Scripting File
- PWSCF files:
- PWSCF Input File
- PWSCF Output File
- FHI98MD files:
- FHI98MD "inp.ini" File
- FHI98MD "coord.out" File
- Molecular structure files:
- XYZ
- PDB
- Gaussian files:
- Gaussian Z-Matrix File
- Gaussian98 Output File
- Gaussian98 Cube File
Within this cascade menu, the
PWscf's
pw.x Input and Output
files can be opened.
Within this cascade menu, the CRYSTAL-95/98/03 input and properties
files can be opened.
- by the ...Open CRYSTAL-95/98/03
Input menu, the user is able to manipulate standard
geometry input and geometry manipulation keyword sections
(refer to CRYSTAL-95/98/03 User manual) and render the crystal
(molecular) structure.
- the ... Open Crystal-95/98/03
Properties menu reads the
CRYSTAL's
unit 9 (user specifies the filename). User is able to render the
following properties:
- Band widths
- DOS and projected DOS
- band structure (prior to that user can graphically select the
k-path
- various 2D (isolines/colorplanes) and 3D (isosurfaces) plots
(charge density, charge density difference, electrostatic
potential)
This entry is related to WIEN2k GUI. User can choose among:
- Open WIEN2k Struct
File : displays crystal structure from the struct
file.
- Render pre-Calculated
Density : reads struct, output5
and rho files and renders crystal structure and
precomputed charge density.
- Calculate & Render
Density : A 2D or 3D region for charge density
calculation can be interactively selected by mouse-clicking.
XCrySDen generates in5 file(s), calculates and renders charge
density either as isolines/colorplanes (2D) or isosurfaces
(3D).
- Select k-path :
Reads struct file and renders first Brillouin zone with
special k-points. K-path can be selected interactively by
mouse-clicking these points.
- Fermi Surface :
trough a series of tasks user is able to render the Fermi
surface.
Closes the current case. Technically the current
XCrySDen process is terminated and a
new one is started.
Saves currently displayed structure in XSF format.
Saves currently displayed structure and large number of the display
parameters in
XCrySDen
scripting file format. Next time the script will be loaded (i.e. as
xcrysden -s script) the structure will be displayed in
the same way as it was saved, i.e., having the same orientation,
zoom, colors, and other display parameters.
Read more ...
Saves currently displayed structure in the format of
CRYSTAL-95/98/03 input file (active only when used as CRYSTAL96/98
GUI)
Saves currently displayed structure as WIEN2k
struct file.
Prints the currently displayed in EPS format. Please take a look at
this
hint for creating good quality
print-out.
Here the converting program for PPM to various other graphics
format (PNG,GIF,JPG) converion, together with the command line
options can be specified. The
convert program from the
ImageMagick program-package (
http://www.imagemagick.org/) is a
very convenient choice.
Read More ...
Periodic Table of Elements can be found here.
In this cascade menu one can found example files included in the
XCRYSDEN distribution for all supported file formats.
Terminates the application.
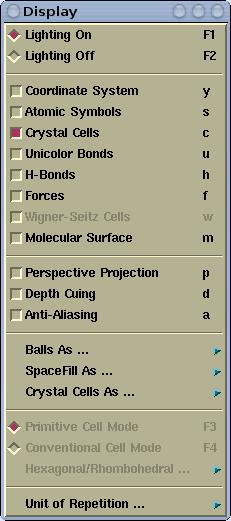
Display menu has the following items:
Toogles between lighting on and off mode
Toggles the display of XYZ Cartesian coordinate system, which is
rendered at the lower left corner of the render window.
Toggles the display of atomic symbols.
Toggles the display of crystal cells (only for periodic structures:
polymers, slabs, crystals).
Toggles the display between the bonds that have the same color as
atoms and so-called
unicolor bonds where all the bonds
have the same color.
Toggles the display of H-bonds. See also the
Modify-->H-bonds
settings menu.
Toggles the display of forces, which are rendered as vectors. The
settings of the force display can be done via
Modify-->Force Settings menu.
Toggles the display of Wigner-Seitz cells. The settings of the
Wigner-Seitz cell display can be done via
Modify-->Wigner-Seitz Cells Settings
menu.
Toggles the display of
Molecular surface. The settings
of the Molecular surface display can be done via
Modify-->Molecular Surface Settings
menu.
Toggles between perspective and orthographic projections.
Makes more distance object appear "darker" (or more foggy). This
effect produces more realistic appearance of the scene. The
depth-cuing can be customized via the
Material/Fog/Antialias Parameters menu.
The images displayed on the screen are aliased due to final
resolution of the screen (typically this is 75dpy). One can improve
this aliasing by a technique called anti-aliasing. It enhances the
graphics quality. Anti-aliasing is computationally very demanding,
hence it typical use is for printing, where it is very useful,
because it makes much superior print-out. The anti-aliasing can be
customized via the
Material/Fog/Antialias Parameters menu.
-->Balls based on
covalent radii
-->Balls based on van der Waals
radii
Switches between covalent and Van der Waals radii for the balls
(applies to BallSticks display mode). Ball size is calculated
as:
ball_size = ball_factor * spacefill_factor *
atomic_radius.
-->Balls based on
covalent radii
-->Balls based on van der Waals
radii
Switches between covalent and Van der Waals radii for the spacefill
sphere (applies to SpcaeFill display mode). Spacefill size is
defined as:
spacefill_size = spacefill_factor * atomic_radius.
-->Display crystal cells
as lines when lighting
-->Display crystal cells as rods when
lighting
In
Lighting-On mode crystal cells can be rendered as lines
or as rods (sticks) with shades (similar to bonds). Switches
between lines and rods display-mode. This option has no effect in
Lighting Off mode.
Toggles between the display of primitive or conventional cell-mode.
-->Unit cell
-->Translational asymmetric unit
Switches between two possible display-modes of the unit cell. What
is meant by this?
Simply rendering just atoms belonging to the zero
reference unit cell would not appear very nice. Why? Let us take
for example an fcc unit cell. The atoms belonging to this
cell have the following crystal (i.e. fractional) coordinates:
(0,0,0); (1/2,1/2,0); (1/2,0,1/2); (0,1/2,1/2). If solely this
atoms are rendered they appear as shown on the left of the below
figure. Instead, what one really wants is the shown on the right of
the figure:


The content displayed on the left of the figure is called
Translational asymmetric unit, while the one on the right
is called "nicely cut unit cell" or shortly Unit
Cell.
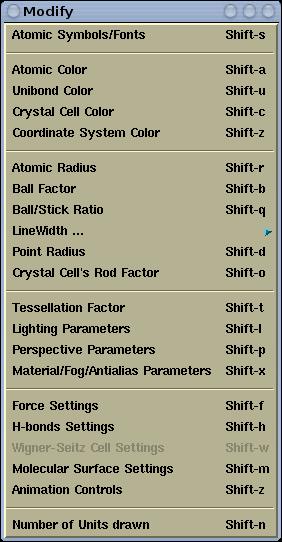
Modify menu has the following items:
Atomic symbols can be customized (edited). Different fonts and
colors can be assigned to different labels.
Read more ...
Pops-up toplevel window, where colors for individual elements can
be set.
Read more ...
Pops-up toplevel window, where color for
unicolor bonds
can be set.
Pops-up toplevel window, where color for crystal cells can be set.
Pops-up toplevel window, where the color of the coordinate system
ban be set.
Read more
...
Pops-up toplevel window, where chemical connectivity factor,
spacefill factor and covalent radii for individual elements can be
set.
Read more ...
Ball factor can be modified here. Ball factor is used for
calculating the ball sizes (BallStick display-mode). Ball size is
defined as:
ball_seize =ball_factor * spacefill_factor * atomic_radius.
Sets the ball/stick ratio, which determines the thickness of bonds
(sticks) with respect to hydrogen ball size. Thickness of sticks is
defined as:
stick_size = spacefill_factor * ball_factor * ballStick_ratio
* hydrogen_radius
-->WireFrame's
LineWidth Sets the line width for WireFrame
Lighting-Off display mode.
-->PointLines's
LineWidth Sets the line width for PointLines
Lighting-Off display mode.
-->Crystal Cell's
LineWidth Sets the line width for crystal cells (for
line display-mode of crystall cells).
-->Lighting-Off outline
width Sets the outline width for all but wireframe
lighting-off display modes.
-->Lighting-On Wire line
width Sets the line width for the various
lighting-on wires (for example, width of wires for wire display
mode of isosurfaces)
Sets the points size for PointLines Lighting-Off display mode.
Sets the rod factor for crystal cells (for rod display-mode of
crystal cells). Rod thickness is defined as:
rod_thickness = rod_factor *
hydrogen_covalent_radius.
Tessellation factor can be modified here. Tessellation factor
determined the number of tessella that form the spheres, cylinders,
etc. The larger the tessellation factor, the larger the number of
tessella. Using larger tessellation factor makes the molecule to
appear nicer. Use relatively large tessellation factor for printing
(cca. 50).
Read more ...
Here the OpenGL lights can be customized. One can enable up to 6
light sources, position them and set their properties. To set the
lighting parameters is not very easy and intuitive. At least basic
knowledge of OpenGL lighting is required.
Here perspective parameters can be modified. There are three
factors:
fovy,
front, and
back. The smaller
the
fovy the larger the perception of the perspective. With
the
front and
back factors the front and back
clipping planes are set. The smaller the
back parameter the
more the structure (=displayed objects) is clipped from the back
side. The
front parameter is counter-intuitive, meaning the
smaller it is the more the structure is clipped from the front
side.
Here the OpenGL material properties of atoms and bonds can be
customized (Materials). Also the depth-cuing (i.e. fog) and
anti-aliasing parameters can be edited.
Pops-up toplevel window, where the "length" factor for the force
display can be set.
Read more ...
Pops-up toplevel window, where the display of H-bonds can be
configured.
Read more ...
Pops-up toplevel window, where parameters for the display of the
Wigner-Seitz cell can be customized.
Read more ...
Pops-up toplevel window, where molecular-surface parameters can be
customized.
Read more
...
Pops-up toplevel window for controlling the animations. menu is
active only the loaded structure file contains more than one
structure.
Read more ...
Sets the number of displayed unit cells in each
A/B/C
direction.
Read more ...
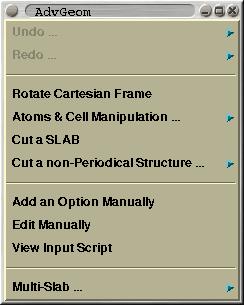
This menu is mented solely for CRYSTAL GUI. Here one can manipulate
an existing structure by various procedures. Among supported
options are: (i) cutting a slab out of a crystal, (ii) generating a
supercell, (iii) adding, removing and substituting atoms, ...
Here you can found
description of this menu and the usage instructions.
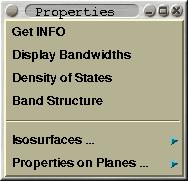
This menu is mented solely for CRYSTAL GUI. Here one can perform
some property analysis. Among supported options are the plotting of
(i) band widths, (ii) density-of-states, (iii) band structure (one
can select th k-path inside Brillouin zone graphically), (iv)
electronic densities and electrostatic potentials as 2D contours or
3D isosurfaces.
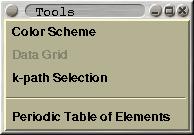
A few useful tools can be found here. This includes:
Color
Scheme, Data Grid, k-path Selection and
Periodic Table of
Elements.
Pops-up toplevel window, where different color schemes can be
selected. Useful when examining the structure (nearest-neighbor
analysis, slab analysis).
If a DATAGRID section (2D or 3D) is present in loaded XSF (XCrySDen
Structure File) then this menu-item is active. Via this option user
will be able to render 3D isosurfaces and/or 2D isolines and
colorplanes.
Read more ...
Renders Brillouin zone with special k-points. K-path can be
selected by mouse clicking these points. So far, selected K-path
can be saved only in CRYSTAL-95/98/03 properties input-file format.
Read more ...
A toplevel window with Periodic Table of Elements pops-up.

![[Figure]](img/xcrysden-picture-small-new.jpg)