XCrySDen program can be
used as a graphical tool for
WIEN2k, a FP-(L)APW program package. The
following graphical tasks can be performed by the
XCrySDen program:
- visualization of crystal structures
- graphical selection of k-path inside the Brillouin zone for
spaghetti plots
- visualization of 2D contours/isolines and 3D isosurfaces
(charge density, electrostatic potentail)
- visualization of Fermi surfaces
All these options are accessible either via
File-->Open WIEN2k ... casade menu or
via command line options.
XCrySDen's command line
options intended for WIEN start with the
--wien_
prefix. Currently five different command line options are
supported:
| Usage: |
xcrysden [OPTIONS]
[file|filehead|directory]
|
xcrysden --wien_struct filehead|file|directory
Corresponding menu:
File-->Open WIEN2k ...-->Open
WIEN2k Struct File Reads struct file and
renders the crystalline structure.
xcrysden --wien_kpath directory Corresponding
menu:
File-->Open WIEN2k ...-->Select
k-path Reads struct file and renders first
Brillouin zone with special k-points. K-path can be selected
interactively by mouse-clicking appropriate k-points. We must
specify EMIN and EMAX parameters and total number
of k-points along the path. This is merely an estimation of the
total number of k-points, since XCrySDen tries to produce a uniform
sampling of k-points along the k-path, therefore don't specify
WIEN2k's maximum allowed number of k-points, as XCrySDen maight generate few points
more.
xcrysden --wien_renderdensity directory
Corresponding menu:
File-->Open WIEN2k ...-->Render
pre-Calculated Density Reads struct,
output5 and rho files and renders crystalline
structure and precomputed charge density.
xcrysden --wien_density directory
Corresponding menu:
File-->Open WIEN2k
...-->Calculate & Render Density First,
either 2D or 3D region for charge density calculation is chosen
grahically by mouse-clicking. Then XCrySDen generates the in5
file(s), calculates and renders charge density. The density can be
displayed either as isolines/colorplanes (2D) or an isosurface
(3D).
xcrysden --wien_fermisurface directory
Corresponding menu:
File-->Open WIEN2k ...-->Fermi
Surface Pops-up a task window for Fermi surface
creation. After several steps the Fermi surface is hopefully drawn
as 3D isosurface. This feature is EXPERIMENTAL, please be careful
!!! So far it was tested on a few spin non-polarised and
spin-polarized systems. (Currently the shift of the k-mesh is not
allowed.)
| LEGEND: |
directory |
name of the case directory
|
filehead |
name of the struct file without
.struct extension
|
filename |
name of the struct file
|
This option is available via
File-->Open WIEN2k ...-->Select
k-path menu or as
--wien_struct command
line option. There is nothing special for this option. Displaying
the crystal structure from WIEN
struct file is alike
displaying the structure from other supported formats. The
decription of various available
XCrySDen options for the
visualization of crystal structures can be found in the following
documents:
Short Introduction to XCrySDen,
Description of XCrySDen main window and menus,
and
HOWTO: Modify Menu.
This option is available via
File-->Open WIEN2k ...-->Select
k-path menu or as
xcrysden --wien_kpath
directory command line option. A special window pops-up
where we select a k-path inside the Brillouin zone (BZ).
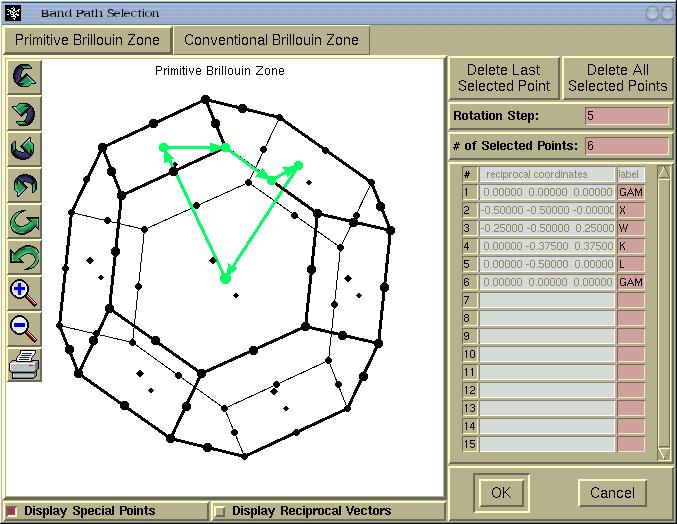
In above window we see two tabs entitled: (i)
Primitive
Brillouin Zone and (ii)
Conventional Brillouin zone.
The latter is provided only for informational purpose, namely, to
see the shape of the BZ tessellated according to the conventional
set of reciprocal vectors. Hence for real applications we stick to
Primitive Brillouin Zone and select a k-path by
mouse-clicking a special k-points. The BZ can be rotated by
holding-down left mouse button and dragging the mouse.
For a few Bravais lattice types, several common k-points will be
labelled automatically (thanks to Peter Blaha), such as GAMMA, X,
W, K, L points for the fcc lattice. The automatic k-point labbeling
currently supports the following Bravais lattice types:
- primitive cubic
- fcc
- bcc
- primitive tetragonal
- body centered tetragonal
- primitive orthorhomobic
When we are done with the k-points selection the [OK]
button should be pressed and a new window will appear.
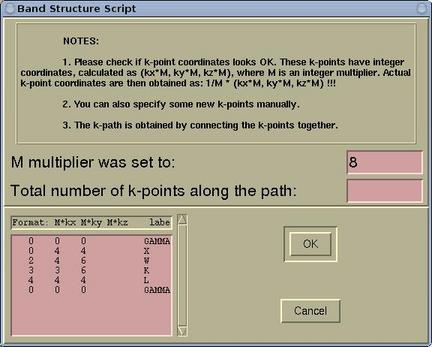
Here we must specify the total number of k-points along the path.
This is merely an estimation of the total number of k-points. The
precise number of k-points is determined by
XCrySDen in such a way that the
density of k-points is as uniform as possible for all k-line
segments. After pressing the
[OK] button the file-browser
will appear and we can save the
.klist file for
spaghetti plot.
This option is available via the
File-->Open WIEN2k ...-->Render pre-Calculated
Density menu or as
xcrysden
--wien_renderdensity directory command line option.
XCrySDen reads the
struct,
output5 and
rho files and renders
crystalline structure and precomputed charge density as contours or
colorplane.
Here you can read how
to control various parameters for contour and colorplane display.
This option is available via the
File-->Open WIEN2k ...-->Calculate & Render
Density or as
xcrysden --wien_density
directory command line option. First
XCrySDen will ask whether we want to
compute the charge density (or some other property) in 2D or 3D
region:
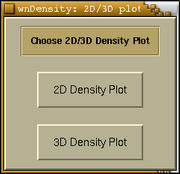
Now we will have to select a region of space where the charge
density will be calculated. Depending on our choice this will be
either 2D or 3D region.
Here you
can read how to select a 2D region, and below you can read how to
select a 3D region.
The following window is devoted to the selection of the 3D region
of space.
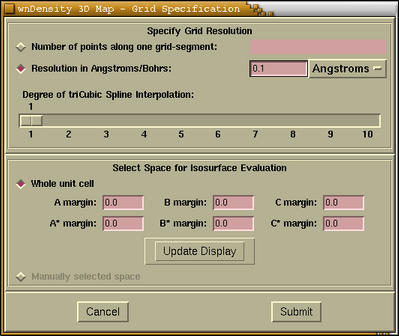
Here is the description of widgets on above window.
| [Grid Resolution] |
You can find the description of this items
here |
| [Interpolation] |
There is a possibility of interpolating a grid
by tri-cubic spline interpolation. At this stage I would suggest to
specify no interpolation (degree=1), as it will be possible to do
that later.
|
| [Space Selection] |
By default the space comprised by the whole
unit cell is selected. Then we have the possibility to specify the
margins. Here you can find the
description about margins. Important: Only after we press
the [Update button] the currently selected space will be
rendered as transparent box. On the figure below we see the
unit-cell space selection with the A=B=C=A*=B*=C*=0.2
margins.
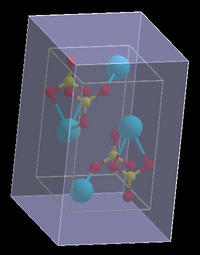
|
There are two buttons on the bottom of
3D Map
- Grid Specification window. The function of these buttons
is the following:
| [Cancel] |
Cancels the process and closes the
window
|
| [Submit] |
Submits the calculation to the WIEN program.
But before that a special window pop-sup, where we specify various
WIEN related flags. ( Read more ...). |
The wollowing window is devoted to the selection of the 2D region
of space.
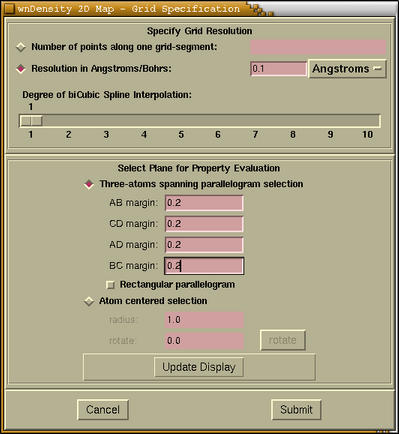
At the top of the window we specify the grid resolution and an
interpolation setting. (
Read more
...). The second part (label
Select Plane for Property
Evaluation) of the
2D Map - Grid Specification window
is devoted to the selection of 2D region of space. We can do that
via two different procedures, which are chosen by pressing the
corresponding radiobutton, that is either
[Three-atoms spanning
parallelogram selection] or
[Atom centered selection].
Here you can find description
of these graphical procedures. Two button are located at the bottom
of
2D Map - Grid Specification window.
The description of their function can be found
here.
In the foolowing window the WIEN flags for charge density
calculation are entered:
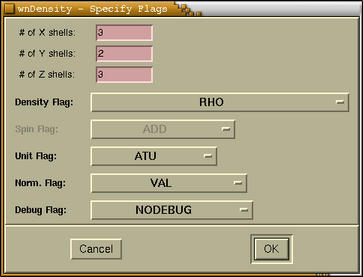
These flags are the same for 2D and 3D charge density calculation,
simply because the latter calculation in composed of several 2D
slice calculation. For the meaning of this flags you should refer
to WIEN manual.
After we have done all above steps then the controlling window for
either contour or isosurface display appears, depending on the 2D
or 3D choice.
Here you can find
description for the contour display, and
here for isosurface display.
This is a new option and was not yet tested extensively, please be
careful!!! It is available via
File-->Open WIEN2k ...-->Fermi
Surface menu or as
xcrysden
--wien_fermisurface directory command line option. First a
task window for Fermi surface creation Pops-up.
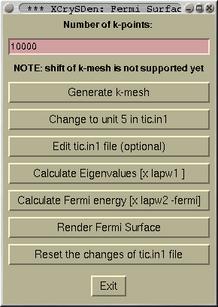
This window style is closely similar to that of
Wien in a
Box, hence it is hopefully self-explanatory. After series of
task (pressing buttons from top to bottom) are performed, one
finally arives to
[Render Fermi Surface button]. Upon
pressing the button the value of Fermi energy will be queried.
After that two windows will appear:
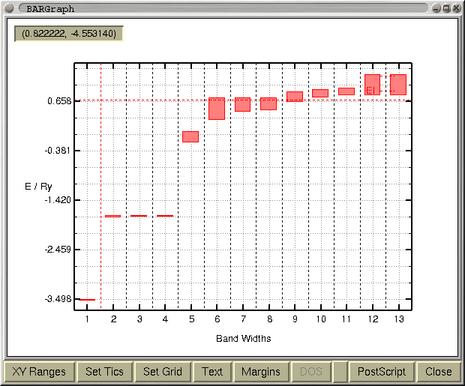
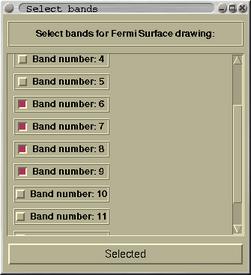
The upper window displays the band-widths. The Fermi level is also
indicated by red horizontal line at 0.66 Ry. The purpose of the
bottom window is the selection of the bands for which the
corresponding part of Fermi surface will be drawn. Usually, one
selects the bands that cross the Fermi level. When the bands are
selected proceed by pressing the button
[Selected]. In a
while the Fermi surface of the first selected band will be
displayed in a viewer window. Actually, the viewer is composed from
notebook, holding the Fermi surfaces of all the bands in separate
pages.
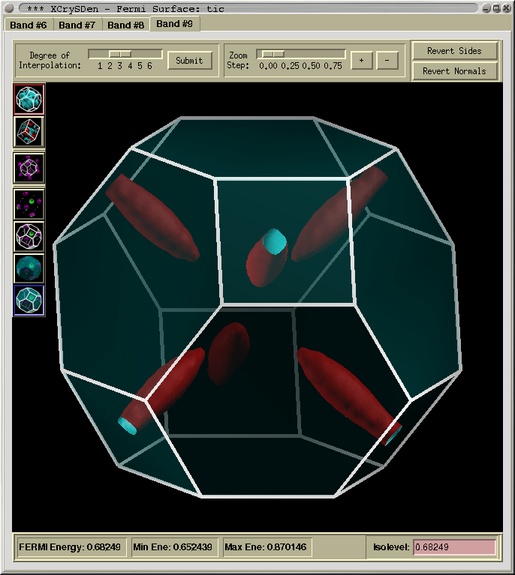
Fermi surface corresponding to the 9th band of TiC (i.e. the
4th band that crosses the Fermi level)
Here you can get more information about
the Fermi surface Viewer ...

![[Figure]](img/xcrysden-picture-small-new.jpg)